
空泡蛋白分类35同源基因(VPS35)突变导致晚发性常染色体显性帕金森病(PD),已知单个错义突变(Asp620Asn, D620N)与PD家族的疾病分离。VPS35基因编码逆转录复合体的核心成分,参与内体分选和跨膜货物蛋白的再循环。VPS35-linked PD在临床上与散发性PD难以区分,尽管尚不清楚VPS35-PD脑是否表现出散发性病例特有的α-突触核蛋白阳性脑干Lewy病理。先前的研究表明,在低等生物中,VPS35与pd相关基因产物α-突触核蛋白之间存在功能相互作用,其中VPS35的缺失增强了α-突触核蛋白诱导的毒性。在小鼠实验中,据报道VPS35过表达可以挽救人α-突触核蛋白转基因小鼠海马神经元的损失,这可能表明这些小鼠存在逆转录酶缺陷。
在这里,我们采用多种完善的遗传啮齿动物模型来探索VPS35和α-突触核蛋白在体内的功能或病理相互作用。
我们发现内源性α-synuclein在病毒介导的小鼠人D620N VPS35诱导的黑质纹状体通路多巴胺能神经变性中是不可缺少的,这表明α-synuclein不作用于VPS35的下游。接下来,我们评估了人类A53T-α-突触核蛋白转基因小鼠受影响脑区的逆转录酶水平,但发现VPS35、VPS26或VPS29的核心亚基水平正常。我们进一步发现,杂合的VPS35缺失并不能改变这些A53T-α-synuclein转基因小鼠的致死性神经退行性表型,这表明在该PD模型中不存在反转录蛋白缺陷。最后,我们在基于病毒的人野生型α-突触核蛋白大鼠PD模型中探讨了增加VPS35表达的神经保护能力。然而,我们发现野生型VPS35的过表达不足以保护α-突触核蛋白诱导的黑质多巴胺能神经变性、α-突触核蛋白病理和反应性胶质瘤。
总的来说,我们的数据表明VPS35和α-突触核蛋白在PD的神经退行性模型中相互作用有限,并且不支持它们在共同的病理生理途径中相互作用。
帕金森病(PD)是一种复杂的神经退行性运动障碍,通常以散发的方式发生,但5 - 10%的病例是遗传和单基因的[1,2,3,4,5]。在家族性PD中,液泡蛋白分选35 (VPS35)基因突变导致晚发性常染色体显性PD[6,7,8]。单个杂合突变Asp620Asn (D620N)已被确定与多个PD家族的疾病明确分离,并且是vps35相关疾病的最常见原因[9]。携带VPS35突变的PD患者表现出与散发性PD难以区分的临床谱和神经影像学表现,尽管尚不清楚这些患者的大脑是否会出现典型的脑干Lewy病理[6,7,8,10,11,12]。阐明VPS35突变导致PD的机制对于定义驱动神经退行性变的常见细胞通路和治疗开发具有重要意义。与其他PD相关基因一起,VPS35的出现突出了内溶酶体途径在PD病理生理中的关键作用[13]。
VPS35编码五聚体反转录复合物的一个核心组分,该复合物在跨膜货物蛋白从核内体向反式高尔基网络或质膜的逆行运输和再循环中起作用[14,15,16,17]。Retromer由一个中心的货物选择性复合物组成,该复合物由VPS35结合到VPS26和VPS29,以及一个在膜结合和变形中起作用的分选连接蛋白二聚体组成[18]。对逆转录物的大部分了解,包括对其选择性货物的识别,都来自于对酵母和哺乳动物细胞系的研究,但对其在脑细胞中的作用仍知之甚少。PD相关的D620N突变如何诱导PD的神经元变性尚不清楚[9,19]。在某些细胞模型中,D620N VPS35可能会影响特定货物的内体分选[20,21,22,23,24],并且通过VPS35与FAM21亚基的相互作用减少,也会损害五聚体WASH复合物向内体的募集[21,23]。在哺乳动物细胞系中,WASH与VPS35结合减少可能导致自噬受体ATG9A的囊泡分选改变和巨噬功能受损[23]。VPS35中的D620N突变也通过调节伴侣介导的自噬受体LAMP2A[22]和线粒体融合/裂变蛋白mitofusin-2或Drp1[25, 26]分别与自噬和线粒体形态的改变有关。
最近的研究表明VPS35与α-突触核蛋白(αSyn)之间存在有趣的关系[22,25,27,28],α-突触核蛋白是PD的家族性和危险基因,也是路易小体的主要成分。在啮齿动物大脑中,有报道称VPS35的杂合性种系缺失或其在多巴胺能神经元中的条件纯合缺失可诱导黑质多巴胺能神经元的缺失,促进αSyn神经元的积累[22,25]。值得注意的是,纯合子种系缺失VPS35会导致早期胚胎致死[29]。D620N VPS35敲入小鼠是一种与PD生理更相关的模型,也表现出多巴胺能神经退行性变,但由于相互矛盾的报道,尚不确定是否存在脑αSyn积累[30,31]。然而,将D620N VPS35敲入小鼠与人A53T-αSyn转基因小鼠杂交并不足以改变这些αSyn小鼠的致死性神经退行性表型[30]。相反,这些敲入VPS35的小鼠在整个大脑中表现出异常tau蛋白的体树突性积累[30]。此外,在成年大鼠黑质纹状体通路中,病毒介导的人D620N VPS35的表达足以诱导黑质多巴胺能神经元丧失,但没有明显的αSyn积累或病理[32]。虽然有一些证据表明VPS35可以调节αSyn积累[22,25,31],但尚不清楚这是否是驱动这些VPS35模型神经退行性变的充分或必要条件。也有证据表明,病理性α - syn可能会导致功能性后转录酶缺乏。例如,VPS35缺失增强了酵母、秀丽隐杆线虫和果蝇PD模型中人αSyn表达诱导的毒性表型[28,33]。慢病毒介导的人野生型(WT) VPS35过表达已被证明可以挽救人WT-αSyn转基因小鼠海马锥体神经元丢失、反应性星形胶质增生和αSyn积累[28]。这些发现支持了αSyn可以诱导逆转录酶缺乏的观点,恢复一个逆转录酶亚基VPS35就足以提供神经保护。报道的VPS35的保护作用是否也与PD的易感神经元群体有关,还需要进一步评估。
在这里,我们开始更好地定义大脑中VPS35和αSyn之间的功能和病理相互作用。我们通过多种啮齿类动物模型,重点研究了αSyn是否在D620N VPS35下游介导小鼠神经退行性变,以及αSyn病理是否可以诱导下游逆转录蛋白缺乏。我们的数据表明,内源性αSyn在D620N vps35诱导的小鼠多巴胺能神经退行性变中是不可缺少的,并且我们没有发现物理或功能上的后转录酶缺乏导致人类αSyn诱导的啮齿动物神经退行性变的证据。我们的研究未能为PD动物模型中VPS35和αSyn之间有意义的或强有力的相互作用提供支持,这表明这些蛋白可能在PD中以独立的途径起作用。
所有动物实验均经范安德尔研究所机构动物护理和使用委员会(IACUC)批准,并严格按照美国国立卫生研究院实验室动物护理和使用协会的规定进行。给啮齿动物随意提供食物和水,光照/黑暗循环12 h,并保持在无病原体屏障设施中。从Charles River实验室获得雌性成年Sprague-Dawley大鼠(体重约180-200 g),用于AAV载体的立体定向递送。VPS35FLOX/WT小鼠携带可破坏VPS35表达的带floxed的“WT迷你基因”插入(Vps35tm1.2Mjff,库存号:021807)从Jackson实验室获得,并在之前进行了描述[30]。SNCA基因敲除小鼠(缺失外显子1-2;Sncatm1Rosl,库存号。003692)和人A53T-α-Syn转基因小鼠(G2-3系,由小鼠朊蛋白启动子驱动;股票没有。006823)已被描述[34]。采用已建立的基因分型方案,通过基因组PCR对小鼠进行鉴定。将半合子人A53T-α-Syn与杂合子VPS35FLOX/WT进行单轮杂交,产生双突变体小鼠(A53T-α-SynTg/+/VPS35FLOX/WT)和相应的同辈对照。
先前曾报道过myc标记的人α-突触核蛋白质粒[35],未标记的人α-突触核蛋白(WT、A30P、E46K、A53T)质粒由Hilal Lashuel教授(瑞士EPFL)获得。一个myc标记的人tau (4R0N亚型,WT)质粒由Leonard Petrucelli教授(佛罗里达州杰克逊维尔梅奥诊所)提供。GIPZ慢病毒质粒共表达turboGFP和mir30适应型短发夹rna靶向VPS35 (shRNA #1,克隆Id: V3LHS_389029)shRNA #2,克隆Id: V2LMM_36638)或Pol II人类CMV启动子的非沉默对照(Dharmacon Cat # RHS4346)从Horizon Discovery获得。
使用的一抗有:小鼠抗v5、抗v5 - fitc、抗v5 - hrp (Life Technologies),小鼠抗myc - hrp(克隆9E10;Roche),小鼠抗vps35(克隆2D3, ab57632;Abcam),兔抗vps26 (ab23892;Abcam),山羊抗vps29 (ab10160;Abcam),兔抗th (NB300-109;Novus Biologicals),小鼠抗α-synuclein(克隆42;BD Biosciences),小鼠抗人α-synuclein(克隆Syn211;Sigma),小鼠抗pser129 -α-synuclein(克隆EP1536Y;Abcam),小鼠抗app(克隆22C11;Millipore),兔抗gfap (ab227761;Abcam),兔抗iba1 (019-19741;Wako),小鼠抗大鼠CD68(克隆ED1;Bio-Rad)和小鼠抗β-微管蛋白(克隆TUB 2.1;σ)。用于Western blotting的酶标二级抗体为:山羊抗小鼠IgG,轻链特异性和小鼠抗兔IgG,轻链特异性(Jackson Immunoresearch)。荧光共聚焦分析使用以下二抗:AlexaFluor-488、-594或-647山羊抗小鼠IgG和AlexaFluor-488或-647山羊抗兔IgG (ThermoFisher)。在明场显微镜下,使用以下生物素化二抗:山羊抗小鼠IgG和山羊抗兔IgG (Vector Labs)。
人SH-SY5Y神经细胞和HEK-293T细胞在添加10%胎牛血清和青霉素/链霉素的Dulbecco 's modified Eagle 's medium (DMEM) (Life Technologies)中,37℃、5% CO2保存。使用XtremeGene HP DNA转染试剂(Roche)根据制造商的说明,用质粒DNA转染细胞,实现瞬时转染。转染后48-72 h收获细胞。为建立稳定表达shRNA的细胞系,将上述pGIPZ-turboGFP/shRNA表达质粒瞬时转染SH-SY5Y细胞,转染后48 h在含嘌呤霉素(2 μg/ml)的培养基中筛选2 - 3周,获得稳定的克隆。
在共免疫沉淀(Co-IP)实验中,将HEK-239T细胞与质粒组合在10 cm培养皿中瞬时共转染。转染后48 h,细胞在1 ml裂解缓冲液(20 mM hepe - koh, pH 7.2, 50 mM醋酸钾,200 mM山糖醇,2 mM EDTA, 0.1% Triton X-100, 1X Complete Mini蛋白酶抑制剂鸡尾酒[Roche Applied Sciences])中收获,在4℃下旋转1小时,并在15,000 rpm下离心15分钟。可溶性部分与预先与小鼠抗v5抗体(2 μg;Life Technologies),在4°C下孵育过夜。Dynabead配合物用1X PBS、0.1% Triton X-100、150 mM NaCl洗涤一次,用1X PBS、0.1% Triton X-100洗涤两次,用1X PBS洗涤三次。在Laemmli样品缓冲液中,95oC煮沸5分钟洗脱IPs。IPs和输入裂解物(总数为1%)通过SDS-PAGE分离,转移到硝化纤维素(0.2 μm;GE Healthcare),用一抗和轻链特异性抗小鼠/兔IgG-HRP偶联抗体进行Western blotting。通过增强化学发光(ECL, GE Healthcare)显示蛋白质,并使用FujiFilm LAS-4000图像分析系统获取数字图像。使用Image Studio Lite (LI-COR Biosciences)对图像进行密度定量。
先前已经描述了将v5标记的人VPS35 (WT或D620N) cdna或填充序列(WPRE)插入pAAV2-mPGK-MCS载体中[32]。重组AAV2/6病毒载体由北卡罗来纳大学病毒载体核心(University of North Carolina viral Vector Core)制作并滴度,如前所述[36]。将病毒稀释至终浓度为1.3 × 1012病毒基因组(vg) / ml。AAV2/6-PGK-α synt - wt - wpre或AAV2/6-PGK- mcs - wpre载体由Bernard Schneider博士(Bertarelli Foundation Gene Therapy Platform, EPFL, Switzerland)提供,并在之前的文献中描述过[37],将其稀释至病毒滴度为1 × 1010转导单位(TUs) / ml。
如前所述进行立体定向注射[32,36,37]。简单地说,3-4月龄的SNCA KO或WT窝代小鼠单侧注射AAV2/6-VPS35或AAV2/6-MCS-WPRE载体进入黑质,相对于bregma的坐标如下:前后(A-P), -2.9 mm;中外侧(M-L), -1.3 mm;背-腹侧(D-V), -4.2 mm。每只小鼠以0.2 μl/分钟的流速以2 μl的体积注射病毒滴度为~ 2.6 × 109 vg的AAV2/6。注射后12周处死动物。
成年女性Sprague-Dawley老鼠(180 - 200 g)收到一个单边intranigral熔池下面的重组AAV2/6向量与转基因表达无处不在的PGK1强启动子的控制下:(1)AAV-PGK-MCS-WPRE x2, (2) AAV-PGK -αSyn-WT-WPRE + AAV-PGK-MCS-WPRE, (3) AAV-PGK-VPS35-WT + AAV-PGK-MCS-WPRE, (4) AAV-PGK-VPS35-D620N + AAV-PGK-MCS-WPRE, (5) AAV-PGK -αSyn-WT-WPRE + AAV-PGK-VPS35-WT, (6) AAV-PGK -αSyn-WT-WPRE + AAV-PGK-VPS35-D620N。AAV-αSyn-WT-WPRE或AAV- mcs - wpre载体与AAV- vps35或AAV- mcs - wpre载体的滴度为~ 1 × 107转导单位(TUs),滴度为~ 2 × 109 vg。在大鼠黑质内单侧注射总容积4 μl病毒,坐标为:A- p, -5.2 mm;M-L, -2.0 mm;D-V,?7.8 mm(相对于bregma)。注射后14周处死动物。
在6倍体积的Triton裂解缓冲液(50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5%甘油,1% Triton X-100, 1 mM EDTA, 1X Complete Mini蛋白酶抑制剂混合物[Roche])中快速显微解剖和均质。使用机械均质机(IKA T10 basic, Ultra Turrax)破坏组织,在4°C下10万x g超离心30分钟后获得triton可溶性部分。在3体积的SDS裂解缓冲液(50 mM Tris-HCl, pH 7.4, 2% SDS, 1X Complete Mini蛋白酶抑制剂鸡尾酒[Roche])中进一步超声提取微球,并在25°C下21,000 x g离心30分钟,获得SDS可溶部分(triton -不溶部分)。采用BCA法(Pierce Biotech)测定蛋白浓度。
采用0.1 M磷酸盐缓冲液(pH 7.3)经心灌注4%多聚甲醛(PFA)。取脑,4% PFA固定90 min, 30%蔗糖冷冻过夜,制作40 μm厚的冠状切片。为了进行显色免疫染色,切片在4°C下用甲醇稀释的3% H2O2 (Sigma)孵育10分钟,以淬灭内源性过氧化物酶活性。切片用10%正常山羊血清(Invitrogen), 0.1% Triton-X100在PBS中阻断,室温下1小时。切片与一抗在4℃下孵育48 h,与生物素化二抗(Vector Labs)在室温下孵育2 h。ABC试剂(Vector Labs)室温孵育1小时后,在3,3 ' -二氨基联苯胺四盐酸(DAB)中可视化;Vector Labs),切片安装在Superfrost plus载玻片(Fisher Scientific)上,用增加乙醇浓度和二甲苯脱水,并用Entellan(默克)盖盖。所有图像均使用光学显微镜(Axio Imager M2,蔡司)和彩色CCD相机(AxioCam,蔡司)进行拍摄。
在室温下,用10%正常山羊血清(Invitrogen)、0.4% BSA和0.2% Triton-X100在PBS中阻断切片1小时,进行荧光免疫染色。切片用一抗在4℃下孵育48 h,二抗偶联到AlexaFluor-488、AlexaFluor-594或AlexaFluor-647 (Life Technologies)在室温下孵育2 h。切片在Superfrost plus载玻片(Fisher Scientific)上切片,用含有DAPI (Invitrogen)的extend载玻片盖盖。使用配备100倍油物镜的尼康A1plus-RSi激光扫描共聚焦显微镜(尼康仪器)捕获图像。
如先前所述,使用StereoInvestigator软件(MicroBrightField Biosciences)的光学分分器探针,对黑质致密部th阳性多巴胺能神经元和nnisl阳性神经元的数量进行无偏立体估计[30,32,37]。每隔4个连续冠状切片取40 μm厚度,覆盖整个黑质区,用抗th抗体免疫染色,用甲酚紫(Nissl)染色反染。对于小鼠,采用系统随机方式取样,采用覆盖黑质的120 × 120 μm正方形网格,并使用由50 × 50 × 20 μm方长方体组成的光学解剖器。大鼠采用220 × 240 μm正方形网格覆盖黑质。研究人员对每种基因型进行盲法分析。
采用分辨率为0.5 μm/像素的ScanScope XT切片扫描仪(Aperio),在20倍放大率下捕获40 μm厚切片的数字图像。如前所述,使用HALO分析软件(Indica Labs Inc.)中的Area Quantification和Microglial Activation模块手动进行图像定量[38,39]。每种免疫组织化学染色(黑质或纹状体TH中的pSer129-αSyn, GFAP, Iba1和CD68)的分析阈值都进行了优化,以便在不包括背景染色的情况下,对高密度和低密度病理区域进行广泛的病理检测。对同侧和对侧黑质或纹状体的组织切片进行手工勾画,并使用阳性像素算法对不同免疫染色所占的阳性染色百分比面积进行量化,每只动物随机选择2-4张图像进行采样。不同免疫组织化学标记病理的阳性染色被设置为像素强度的基线阈值,这样任何像素在该强度或更大(即较暗)被量化为像素阳性区域。每个部分的每个区域轮廓的正像素数被归一化,以考虑轮廓区域到区域的区域可变性。在Halo中使用相同的参数批量分析所有切片/图像。
为了开始探索VPS35和αSyn之间的功能关系,我们通过共免疫沉淀(co-IP)在HEK-293T细胞中短暂共表达v5标记的VPS35变体和未标记的或myc标记的αSyn,评估了这两种蛋白的相互作用。通过Western blot分析,人类VPS35野生型(WT)或pd连锁变体(P316S, D620N)的IP无法检测到与人类WT αSyn的强大相互作用(图1A- b),而VPS35变体确实与内源性逆转录亚基VPS26相互作用(图1A)。先前的研究表明αSyn过表达可诱导逆转录酶缺陷[28]。为了评估这种可能性,我们在人SH-SY5Y细胞中短暂表达了未标记的人αSyn变异体。Western blot分析显示,αSyn的WT或pd相关变体(A30P、E46K、A53T)的过表达不会改变内源性逆转录亚基VPS35、VPS26或VPS29的稳态水平(图1C-D)。既往研究也表明,VPS35耗竭可诱导αSyn的积累[22,25,28]。为了验证这一想法,我们生成了SH-SY5Y细胞克隆,稳定表达mir30适应的短发夹rna (shRNAs),靶向VPS35或非沉默对照,具有或不具有未标记的人类WT αSyn的短暂过表达。两种不同的针对VPS35的shrna成功地显著降低了内源性VPS35蛋白的水平,也诱导了VPS26和VPS29的相应降低(图1E)。VPS35缺失不能增加SH-SY5Y细胞中内源性αSyn蛋白的水平,但令人惊讶的是,会导致过表达WT αSyn水平的降低(图1E)。在这些短暂表达myc标记的人tau的稳定shRNA细胞中进行的类似实验也揭示了VPS35缺失诱导的tau蛋白水平降低(图1F),这很可能表明VPS35-shRNA细胞相对于非沉默shRNA细胞表现出较差的转染效率。综上所述,我们的数据最初未能提供以下证据:(i) VPS35与αSyn的生化相互作用,(ii) αSyn诱导的逆转录酶缺乏,以及(iii) VPS35消耗诱导αSyn积累在人类细胞中。
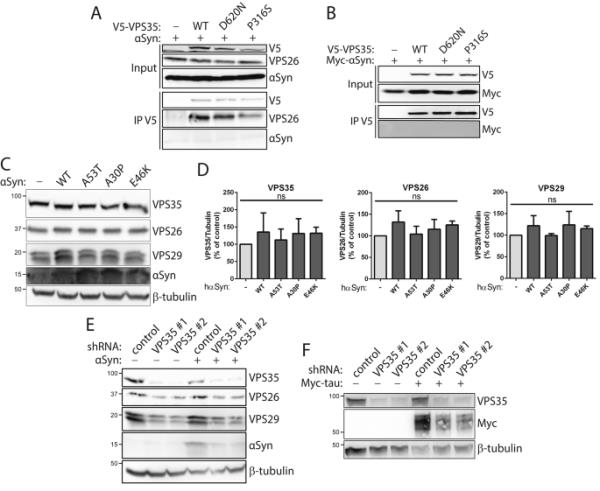
αSyn与VPS35在人细胞中缺乏相互作用。A-B) v5标记的人VPS35 (WT, D620N和P316S)与(A)未标记或(B) myc标记的人WT αSyn之间的Co-IP测定表明VPS35与αSyn缺乏相互作用。将共表达V5标记的VPS35 (WT、D620N或P316S)和WT αSyn的HEK-293T细胞提取物用抗V5抗体进行IP处理,并在IP和输入部分检测αSyn、Myc (αSyn)、VPS26和V5。VPS35变体与内源性VPS26的相互作用等效,但与αSyn的相互作用不等效。C-D) PD-linked αSyn变异体的过表达不影响内源性反转录亚基的稳态水平。C-D) Western blot分析瞬时表达未标记的人αSyn变异体(WT, A53T, A30P或E46K)或空载体的SH-SY5Y细胞的tryon可溶性提取物。图表显示每个后转录亚基VPS35、VPS26或VPS29的水平归一化为β-微管蛋白水平(平均值±SEM, n=3个实验)。数据分析采用单因素方差分析和Dunnett事后分析。ns,非重要。E)内源性VPS35的敲低不会增加内源性或过表达的人WT αSyn的水平。Western blot分析SH-SY5Y细胞克隆的triton可溶性提取物稳定表达mir30适应shRNA靶向VPS35(#1或#2)或非沉默对照shRNA。在表达VPS35- shrnas的细胞中,VPS35水平明显降低,VPS26和VPS29也相应降低,而过表达的αSyn也降低。F)对稳定表达VPS35-shRNAs的SH-SY5Y细胞进行Western blot分析,发现myc标记的人WT tau过表达量也有类似的减少
目前尚不清楚携带VPS35突变的PD脑是否与Lewy病理相关[7],并且D620N VPS35敲入小鼠模型似乎不表现出αSyn聚集随着年龄的增长[30]。由于我们的细胞系实验没有发现VPS35和αSyn之间的相互作用,我们转向啮齿动物模型,首先评估内源性αSyn是否在功能上需要介导PD-linked D620N VPS35的致病作用。我们将表达v5标记的人D620N VPS35(或含有填充序列MCS的对照病毒)的重组AAV2/6载体单方传递到年龄匹配的成年纯合子SNCA (αSyn)敲除(KO)或WT幼崽小鼠的黑质。我们之前已经证明,在成年小鼠和大鼠中,病毒介导的D620N VPS35介导的黑质多巴胺能神经元进行性损失约30%[32,36]。在小鼠注射后12周,我们观察到D620N VPS35的强表达仅限于注射的WT和SNCA KO小鼠的同侧黑质致密部,并且与多巴胺能神经元标记物酪氨酸羟化酶(TH)基本共定位(图2A)。为了评估αSyn缺失对神经退行性变的影响,采用无偏体视学方法计算同侧(注射)黑质与对侧(未注射)黑质中th阳性多巴胺能神经元的数量和nissl阳性神经元的总数。D620N VPS35的表达在WT小鼠中诱导黑质多巴胺能神经元的显著丧失,而在KO小鼠中没有显著改变,相对于对照病毒的影响可以忽略不计(图2B-C,图S3A)。在所有条件下,TH阳性神经元的损失与nsil阳性神经元的损失相当(图2C,图S3B),证实了多巴胺能神经元的变性,而不是TH表型的损失。D620N VPS35的表达也引起纹状体中th阳性多巴胺能神经末梢的显著但不显著的损失,但在WT和KO小鼠之间没有差异(图2D-E)。我们的数据表明,内源性αSyn在小鼠黑质纹状体途径中由病毒介导的D620N VPS35表达诱导的多巴胺能神经变性中不是必需的。
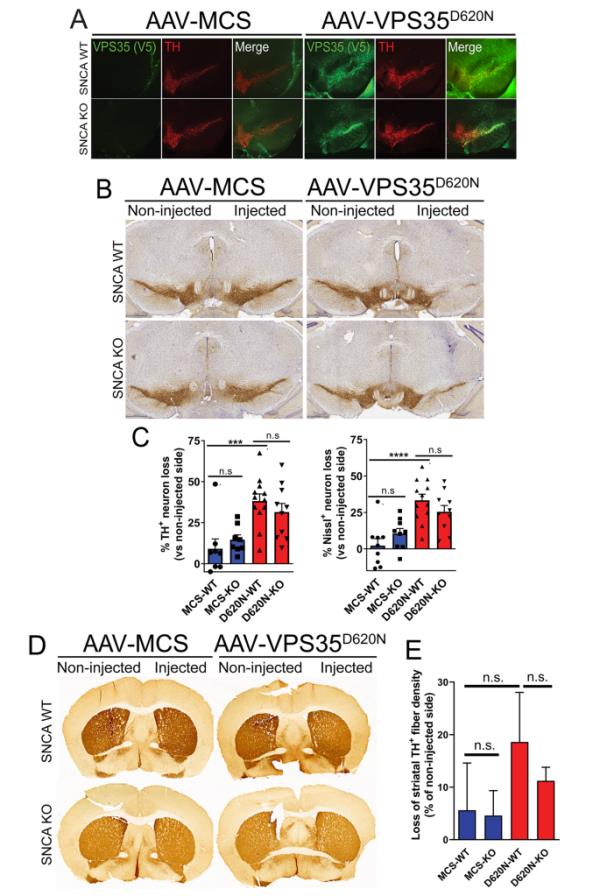
人D620N VPS35表达诱导小鼠多巴胺能神经变性不依赖内源性αSyn。A)注射AAV2/6载体(对照MCS或D620N VPS35) 12周后,WT或SNCA KO小鼠同侧黑质中人VPS35 (V5)与th阳性神经元的免疫荧光共定位。B)单侧黑质内注射MCS或D620N VPS35载体12周后黑质th阳性神经元的免疫组化染色。C) 12周时D620N VPS35或对照病毒诱导的WT和SNCA KO小鼠黑质th阳性多巴胺能和总nsll阳性神经元损失的体视学定量。数据以相对于未注射黑质的神经元损失百分比表示,条形图表示平均值±SEM, n=9-12只小鼠/组。***经Tukey多重比较检验的单因素方差分析P < 0.001或****P < 0.0001,如所示。D) AAV载体递送后12周纹状体th阳性神经末梢免疫染色的代表性显微照片。E)光密度定量纹状体th阳性纤维。数据以th阳性纤维相对于未注射侧的损失百分比表示,条形图表示平均值±SEM, n=9-12只小鼠/组。经Tukey多重比较检验的单因素方差分析,不显著
接下来,我们使用免疫组织化学分析来评估D620N VPS35在小鼠黑质中的表达是否足以诱导经典神经病理标志物的改变。相对于对侧黑质,D620N VPS35的表达(而不是对照病毒)诱导了注射后的WT小鼠黑质中磷酸化ser129 -αSyn (αSyn的一种病理形式)的定性积累(图3A)。D620N VPS35无法在SNCA KO小鼠中诱导磷酸化ser129 -αSyn免疫染色,从而证实了该αSyn标记物的特异性(图3A)。值得注意的是,WT小鼠黑质中D620N VPS35的表达并未改变总αSyn的免疫反应性(图3A),而SNCA KO脑缺乏总αSyn的信号。我们还评估了淀粉样前体蛋白(APP)的免疫染色,APP是轴突损伤的敏感标志物[30,40]。有趣的是,与未注射黑质相比,D620N VPS35的表达增加了WT小鼠注射黑质中细胞体细胞和神经突的APP阳性染色,而在SNCA KO小鼠中,APP染色的增加明显减弱(图3A)。这表明D620N VPS35诱导的APP积累增加依赖于内源性αSyn。
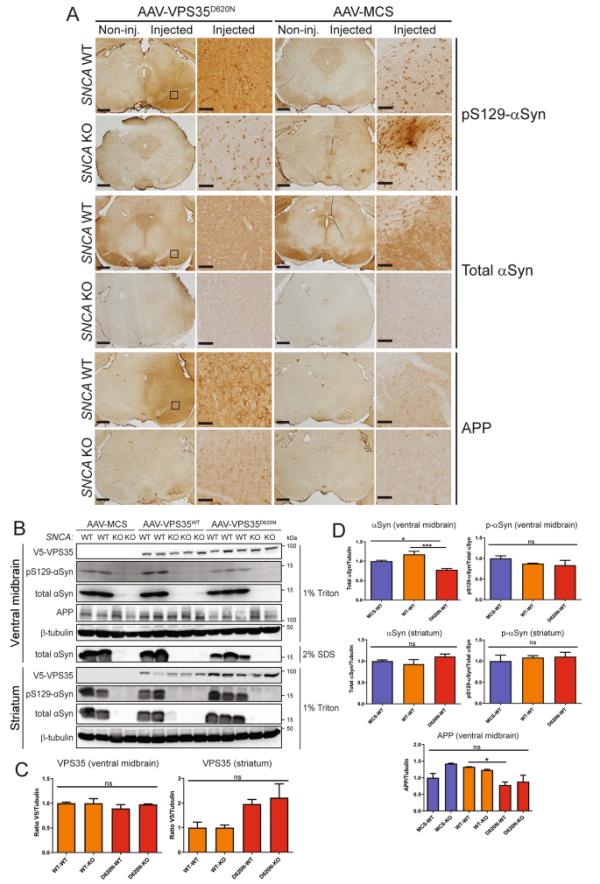
D620N VPS35表达诱导WT小鼠黑质中pSer129-αSyn和APP的积累A)在输注表达人D620N VPS35的AAV2/6载体(相对于MCS) 12周后,WT和SNCA KO小鼠的黑质免疫组织化学染色显示pSer129-αSyn和APP在WT而非KO小鼠中特异性积累。总αSyn水平/分布在WT小鼠中正常,但在KO小鼠中缺失。所示为注射黑质(方框)的高倍图像。B) Western blot分析注射AAV2/6载体(MCS, WT VPS35或D620N VPS35)后12周,WT或SNCA KO小鼠的腹侧中脑和纹状体提取物(1% Triton-X100或2% SDS馏分)。用V5 (VPS35)、pS129-αSyn、总αSyn和APP抗体进行印迹检测,以β-微管蛋白为负载对照。C-D) WT或KO小鼠腹侧中脑和纹状体提取物中人VPS35 (V5)、总αSyn、pS129-αSyn和APP水平的密度分析。pS129-αSyn水平与αSyn总水平归一化。柱状图表示平均值±SEM (n=3-4只动物/组)。*P < 0.05或***P < 0.001经单因素方差分析和Dunnett事后分析,如所示。非标准n。
为了进一步评估这些标记的变化,并确认这些动物中人类VPS35变异的等效水平,在注射后12周,对注射的腹侧中脑和纹状体的triton可溶性或sds -可溶性提取物进行了Western blot分析。在这里,我们还提供了表达人WT VPS35的AAV2/6载体进行比较。WT和D620N VPS35在WT和SNCA KO小鼠的triton可溶性腹侧中脑提取物中检测到相同水平(图3B-C)。在海藤可溶性纹状体提取物中也检测到WT和D620N VPS35,从而证实了人VPS35从注射的黑质到WT和KO小鼠纹状体的有效顺行轴突运输(图3B-C)。与对照病毒相比,WT或D620N VPS35在WT小鼠的triton -可溶性腹侧中脑或纹状体提取物中表达的总αSyn和pSer129-αSyn水平没有增加,并且在KO脑中不存在(图3B, D,图S4A)。相反,我们观察到相对于对照病毒或WT VPS35, D620N VPS35诱导的腹侧中脑总αSyn水平适度下降(图3B, D)。在表达VPS35变体的triton可溶性腹侧中脑提取物中,全长APP水平也没有增加,相反,D620N VPS35相对于WT蛋白显著降低了WT小鼠的APP水平(图3B, D)。虽然我们的生化数据不支持D620N VPS35在WT小鼠中诱导pSer129-αSyn或全长APP蛋白的积累(图3B-D),但免疫组织化学分析(图3A)表明,这些数据可能表明这些病理标记物在中脑腹侧神经元中以α syn依赖的方式重新分布。因此,D620N VPS35在小鼠黑质纹状体通路的表达不产生明显的αSyn神经病理,这与αSyn的病理聚集一致。
最近的研究表明,逆转录物在调节表达人WT αSyn的转基因小鼠海马αSyn的积累以及病毒介导的VPS35过表达在该模型中具有神经保护作用[28]。这些研究提示病理性α - syn可能通过诱导逆转录酶缺乏介导神经毒性。为了探索这种可能性,我们首先评估了朊病毒启动子(PrP)驱动的人类A53T-αSyn转基因小鼠的逆转录酶水平,这些小鼠出现了进行性神经退行性表型,其特征是脊髓和脑干神经元变性、反应性胶质增生、αSyn聚集和运动缺陷,导致肢体瘫痪和过早死亡(7至15个月)[34]。通过Western blot分析,我们评估了6个月大无症状(图4A)或终末期症状(图4B-C) A53T-αSyn小鼠及其非转基因幼崽四个受影响脑区的核心反转录亚基VPS35、VPS26和VPS29的稳态水平。我们发现6月龄和终末期A53T-αSyn小鼠的脊髓、脑干、腹侧中脑或纹状体的后转录酶水平没有显著改变(图4A-C)。正如预期的那样,在A53T-αSyn小鼠的每个脑区都可以检测到人αSyn和pSer129-αSyn的表达(图4A-B)。我们的数据未能提供证据证明A53T-αSyn小鼠随着年龄的增长,受影响的大脑区域中核心逆转录子亚基的耗损。
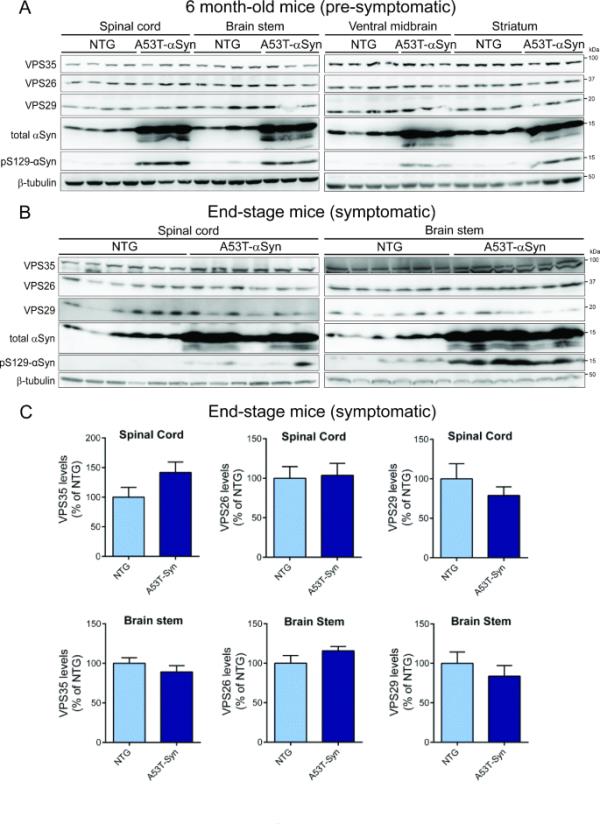
人A53T-α-Syn转基因小鼠脑内不存在逆转录酶缺乏。A) 6月龄症状前半合子A53T-αSyn转基因小鼠及其非转基因窝仔(n=3-4只/组)脊髓、脑干、腹侧中脑和纹状体triton可溶性提取物中反转录亚基和αSyn(总或pS129)的Western blot分析。B)对有症状的终末期半合子A53T-αSyn小鼠(~ 12-13个月)及其非转基因窝仔(n=6只/组)脊髓和脑干提取物进行类似的Western blot分析。C)终末期A53T-αSyn转基因和非转基因小鼠脊髓和脑干中VPS35、VPS29或VPS26水平的密度分析。数据以非转基因小鼠的百分比表示,条表示平均值±SEM (n=6只动物/组)。非配对双尾学生t检验不显著
为了进一步探讨α syn诱导的神经毒性是否介导或加剧逆转录酶缺乏,我们将杂合的VPS35缺失小鼠与A53T-αSyn转基因小鼠杂交。我们之前描述了携带条件等位基因(固定的“WT VPS35”迷你基因)的活杂合VPS35FLOX/WT小鼠,该等位基因无意中破坏了正常的VPS35表达,并作为空等位基因[30]。通过Western blot分析,我们发现VPS35的杂合性不会改变终末期A53T-αSyn小鼠纹状体、腹侧中脑、脑干或脊髓中pSer129-αSyn或总αSyn的稳态水平(图5A-B)。VPS35以及VPS26和VPS29的水平在VPS35FLOX/WT大脑中显著降低(图5A, C),正如预期的那样。我们还使用pSer129-αSyn抗体检测了6个月大症状前小鼠的黑质αSyn病理,然而,VPS35杂合性并没有定量地改变A53T-αSyn小鼠中pSer129-αSyn阳性病理的分布、形态或负荷(图5D-F)。如预期的那样,缺乏A53T-αSyn转基因的VPS35WT/WT和VPS35FLOX/WT小鼠没有表现出αSyn病理(图5D-F)。观察VPS35杂合性对A53T-αSyn小鼠24个月生存率的影响。我们发现A53T-αSyn小鼠在10 ~ 18个月的时间内由于神经退行性变而出现生存受损和过早死亡,去除一个VPS35等位基因没有显著效果(图5G)。最后,我们进行了初步研究,以探讨VPS35杂合性是否影响单侧胃内递送小鼠αSyn预形成原纤维(PFFs)诱导的αSyn病理的初始发展。在接种PFFs后30天,我们在与th阳性多巴胺能神经元共定位的同侧黑质致密部中检测到pSer129-α syn阳性病理,但VPS35WT/WT和VPS35FLOX/WT小鼠的病理程度在质量上相似(图S1)。我们的数据表明,逆转录酶缺乏并不影响A53T-αSyn转基因小鼠的致死性神经退行性表型,也不会影响pff小鼠模型中αSyn病理的初始传播。
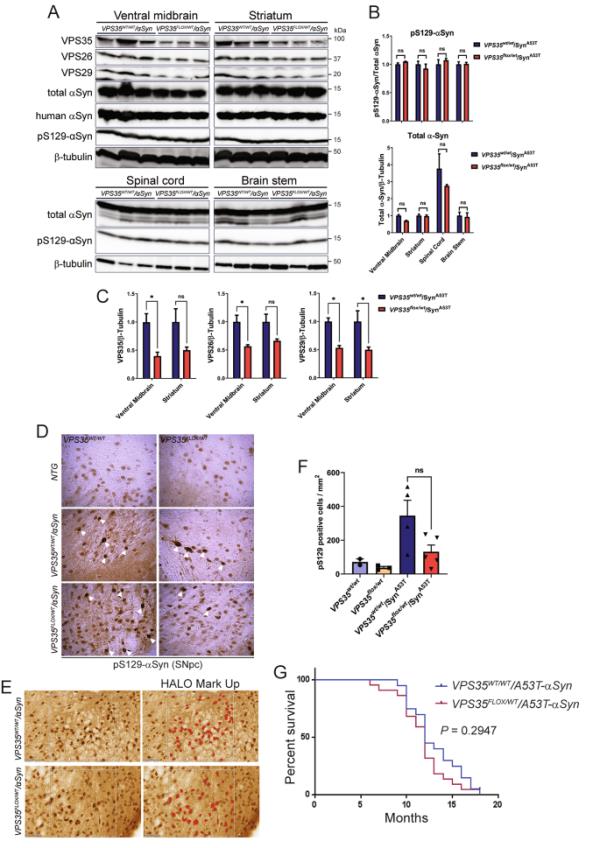
杂合缺失VPS35不能改变人A53T-α-Syn转基因小鼠的α-Syn水平和病理或过早存活。A) Western blot分析约13月龄杂合子VPS35小鼠与人A53T-α-Syn转基因小鼠(VPS35FLOX/WT/αSyn)与A53T-α-Syn同胎小鼠(VPS35WT/WT/αSyn)杂交后腹侧中脑、纹状体、脊髓或脑干1% tritonsoluble提取物。印迹检测逆转录亚基(VPS35、VPS26和VPS29)、总(Syn1)、人(Syn211)和病理(pS129-αSyn) α-synuclein或β-微管蛋白。B) pS129-αSyn归一化为总αSyn或总αSyn归一化为β-微管蛋白的密度分析,以VPS35WT/WT/αSynA53T小鼠的比例表达(平均值±SEM, n=3只小鼠/基因型)。经Bonferroni事后检验的单因素方差分析P > 0.05,如所示。非重大的n。C) VPS35、VPS29或VPS26在中脑腹侧和纹状体中归一化至β-微管蛋白水平的密度分析(平均值±SEM, n=3只小鼠/基因型)。* p < 0.05;也就是说,非配对双尾学生t检验不显著。D)约6月龄症状前vps335wt /WT/αSynA53T或VPS3FLOX/WT/αSynA53T小鼠黑质(SNpc) pS129-αSyn(白色箭头)免疫组化染色的代表性图像。非转基因(NTG)的同窝小鼠没有表现出预期的特异性α-Syn病理。E)使用HALO分析软件中的对象共定位模块,根据pS129 α-synuclein阳性细胞的大小和形状检测SNpc中pS129 α-synuclein免疫染色的数字病理定量。pS129 α-突触核蛋白病理区显示有或没有分析覆盖(红色)。F)每mm2组织面积pS129 α-突触核蛋白阳性染色细胞总数的定量。柱状图表示平均值±SEM (n=2-5只动物/组)。经Tukey多重比较检验的单因素方差分析,不显著。G)人A53T-α-Syn转基因小鼠的致死性神经退行性表型与VPS35的表达无关。通过对VPS35WT/WT/αSynA53T (n=27)和VPS35FLOX/WT/αSynA53T (n=24)小鼠进行18个月的监测,得出Kaplan-Meier生存曲线,直到动物因终末期疾病发作而不得不安乐死。经log-rank (Mantel-Cox)检验,两种基因型患者的生存率无显著差异(P=0.2947)。
接下来,我们试图扩展Dhungel等人最近有趣的观察结果,证明慢病毒介导的人WT VPS35过表达可以减轻人WT-αSyn转基因小鼠的海马神经元丢失和αSyn病理[28]。我们想知道增加VPS35水平是否同样可以对人WT-αSyn诱导的多巴胺能神经变性提供保护。与WT-αSyn转基因小鼠相比,aav介导的人WT-αSyn传递提供了一种更相关的PD啮齿动物模型,因为它概括了黑质纹状体多巴胺能通路的进行性和稳健变性和黑质αSyn病理[37,41,42]。将表达人WT-αSyn和v5标记的人VPS35 (WT或PD-linked D620N)或空对照病毒(MCS)的重组AAV2/6载体单侧共注射到成年大鼠的黑质中。注射后14周,观察多巴胺能神经元变性、αSyn病理及胶质瘤形成。在各动物组同侧大鼠黑质和纹状体中均可通过免疫组化检测到人VPS35变异体和人WT-αSyn(图6A-B),在共聚焦免疫荧光显微镜下,人VPS35变异体和αSyn在th阳性的黑质多巴胺能神经元中基本共定位(图6C)。为了评估VPS35过表达对WT-α syn诱导的神经退行性变的影响,采用无偏体视学方法对黑质th阳性多巴胺能神经元和nissl阳性神经元总数进行计数(图6D-E,图sgc - d)。我们发现,WT-αSyn单独表达(αSyn + MCS)可诱导大鼠同侧黑质多巴胺能神经元损失约57%,而WT VPS35的共表达对这种神经元损失无影响(约54%,图6D-E)。令人惊讶的是,与D620N共表达VPS35可显著降低α syn诱导的神经元损失至33%,但与单独WT-αSyn相比,这种效果并不显著(图6D-E)。在这种较低的病毒滴度下,单独表达WT或D620N VPS35分别诱导约24%或26%的神经元损失(图6D- e),与我们之前的研究相当[32],而空对照病毒产生的神经元损失可以忽略不计(图6D)。通过体视计数,我们观察到nissl阳性的神经神经元总数有相当的损失(图6E,图S3D),证实了多巴胺能神经元变性。WT-αSyn的表达也导致同侧纹状体中th阳性多巴胺能神经末梢的显著损失约20%,这与th阳性神经元的损失相似,但共表达WT VPS35没有显著影响,与D620N VPS35没有显著减少(图6F-G)。综上所述,这些数据表明,在该AAV模型中,WT-αSyn诱导的黑质纹状体多巴胺能神经退行性变中,WT VPS35的表达不足以起到神经保护作用,而D620N VPS35的表达则表现出适度的保护作用。
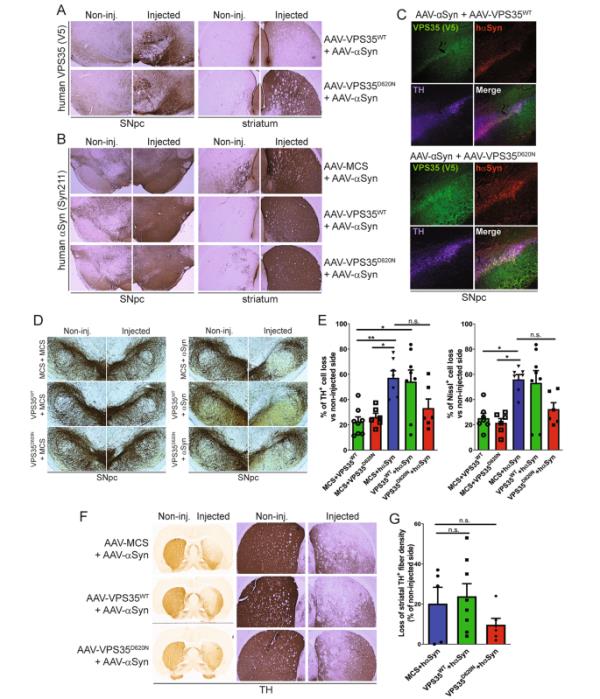
VPS35过表达对人WT-α-Syn诱导的成年大鼠黑质纹状体多巴胺能神经变性无保护作用。A-B) (A)表达WT-α-Syn的AAV2/6载体与VPS35 (WT或D620N)或空对照(MCS)共注射后14周成年大鼠黑质(SNpc)和纹状体中具有代表性的人VPS35 (V5)和人α-突触核蛋白(Syn211)免疫染色的显微照片。显示未注射(左)和注射(右)半球。C)荧光免疫染色显示,注射大鼠黑质14周时,人VPS35 (WT或D620N)和人WT-α-Syn与th阳性多巴胺能神经元共定位。D)联合注射表达人VPS35 (WT或D620N)、人WT-αSyn或空对照(MCS)的AAV2/6载体后14周大鼠黑质(SNpc)抗th免疫染色的代表性显微照片。E)注射AAV载体后14周黑质th阳性多巴胺能神经元和nissl总阳性神经元的无偏立体定量。数据以相对于未注射黑质的细胞损失百分比表示,条形图表示平均值±SEM (n=7-8只动物/组)。经单因素方差分析,*P < 0.05或**P < 0.01。非重大的n。F)注射和未注射AAV载体后14周纹状体th阳性神经末梢免疫染色的代表性显微照片。G)纹状体th阳性免疫染色光密度定量。数据以th阳性纤维相对于未注射侧的损失百分比表示,条形图表示平均值±SEM (n=7-8只动物/组)。经Tukey多重比较检验的单因素方差分析,不显著
接下来,我们试图在该大鼠模型中评估人VPS35表达对αSyn病理和胶质瘤的影响。采用Western blot方法对注射后14周AAV-αSyn/VPS35大鼠腹侧中脑和纹状体提取物进行分析。在中脑腹侧和纹状体的triton -可溶性部分检测到人WT和D620N VPS35蛋白,与D620N蛋白相比,WT VPS35蛋白无显著增加(图7A-C)。与D620N VPS35共表达相对于单独αSyn (+ MCS),这两个脑区中triton -可溶性或sds -可溶性人αSyn的稳态水平没有改变,而WT VPS35共表达导致纹状体中triton -可溶性人αSyn显著增加,中脑腹侧无显著增加(图7A-C,图S4B)。然而,pSer129-αSyn水平在triton可溶性纹状体中与WT VPS35共表达显著降低,而在腹侧中脑中无显著性降低,而D620N VPS35共表达也导致pSer129-αSyn在两个脑区有适度的无显著性降低(图7A-C)。注射后14周,我们利用免疫组化方法进一步评估了VPS35对大鼠黑质αSyn病理和αSyn诱导的反应性胶质瘤的影响。单独注射AAV-αSyn的大鼠在同侧黑质致密部的神经元体和神经突中检测到磷酸化- ser129 -αSyn阳性病理,而未注射黑质的大鼠则没有磷酸化- ser129 -αSyn阳性病理(图8A-B, Q)。使用Halo分析软件定量分析,与单独注射AAV-αSyn相比,D620N VPS35共表达可诱导pSer129-αSyn病理显著增加,而WT VPS35只产生中间作用(图8C-D, Q和图S2)。与未注射对侧黑质相比,单独表达人α - syn不能诱导gmap阳性星形胶质细胞或iba1阳性小胶质细胞占据的面积显著增加,与WT或D620N VPS35共表达也没有影响(图8E-L、R、S和图S2)。我们还使用Halo软件根据iba1阳性小胶质细胞的形态(图8T和图S2)或cd68阳性小胶质细胞(图8M-P, U和图S2)监测激活的小胶质细胞,然而在AAV模型中,人类αSyn单独表达或其与VPS35变体的共表达仅在14周时诱导适度但不显著的小胶质细胞激活。总的来说,这些数据无法令人信服地证明WT VPS35过表达在减轻人αSyn病理或胶质瘤中的神经保护作用。相反,我们观察到WT VPS35诱导的人αSyn水平的生化增加,或者D620N VPS35诱导的pSer129-αSyn阳性病理增加,尽管它对多巴胺能神经变性有保护作用。
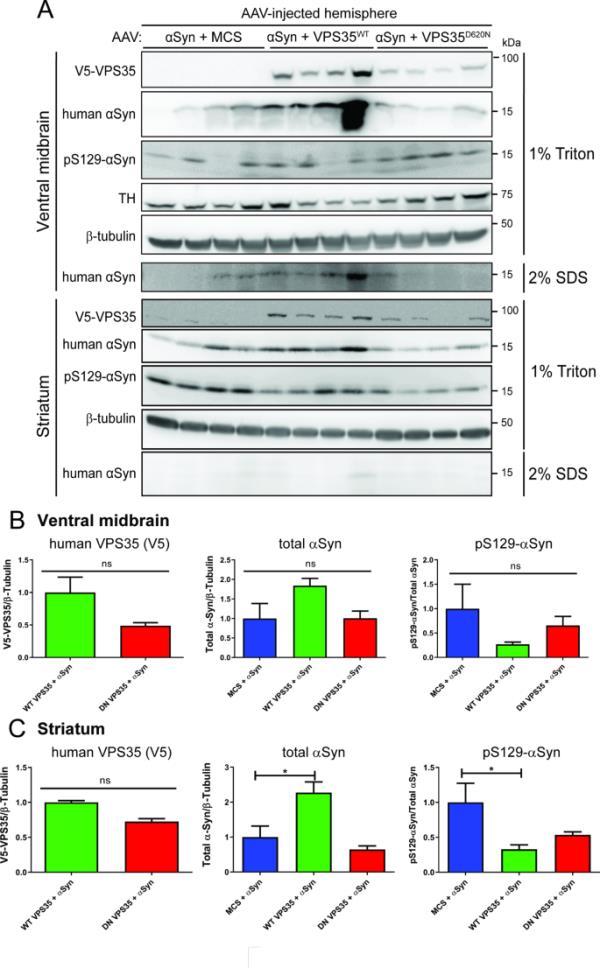
VPS35过表达对人WT-α-Syn水平及大鼠脑磷酸化的生化影响。A)注射表达WT-α-Syn和VPS35的AAV载体(WT或D620N)或空对照(MCS)后14周,Western blot分析大鼠腹侧中脑和纹状体提取物。1% Triton-X100或2% SDS分别用人VPS35 (V5)、人α-Syn (Syn211抗体)、pS129-α-Syn、TH或β-微管蛋白抗体作为对照进行检测。值得注意的是,只有人类WT-α-Syn在腹侧中脑或纹状体的2% SDS中检测到,而pS129-α-Syn或人类VPS35未检测到。B-C) (B)中脑腹侧或(C)纹状体1% tritonsoluble提取物中pS129-α-Syn水平归一化为人α-Syn,或人α-Syn或VPS35 (V5)水平归一化为β-微管蛋白的密度分析。柱状图表示平均值±SEM (n=4只动物/组)。*经Dunnett事后分析的单因素方差分析P < 0.05,如所示。非标准n。
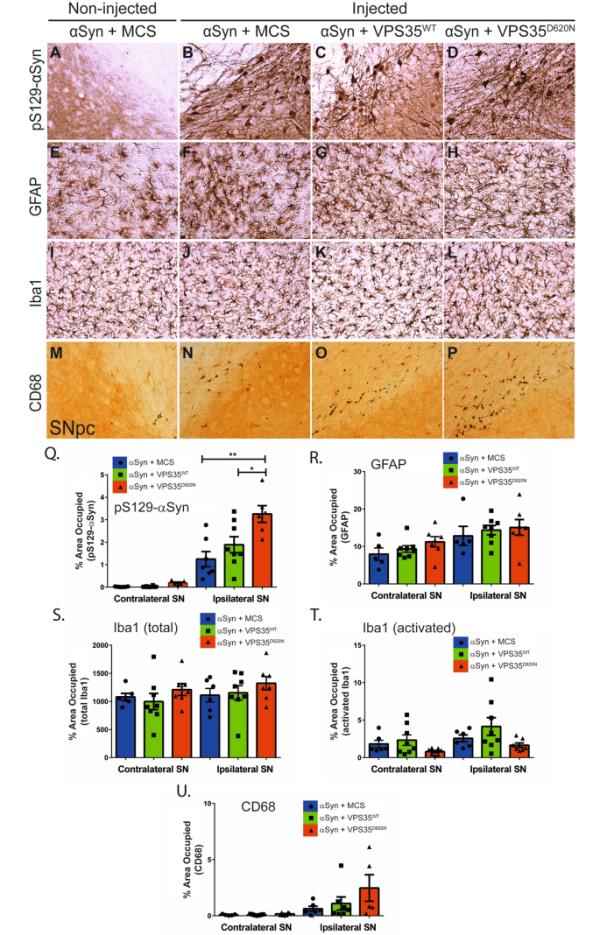
VPS35过表达不会减弱大鼠黑质中人类WT-α- syn依赖的病理。A-P)与人VPS35 (WT或D620N)或空对照(MCS)共注射表达人WT-α-Syn的AAV载体后14周大鼠黑质致密部(SNpc)的免疫组织化学染色。结果显示,注射SNpc对(A-D) pS129-α-Syn、(E-H)星形胶质细胞(gfap阳性)、(I-L)总小胶质细胞(iba1阳性)和(M-P)活化小胶质细胞(cd68阳性)的免疫反应性。图中为α-Syn/MCS对照组未注射SNpc的对比。Q-U)采用HALO分析软件定量各病理标志物。pS129-α-Syn、GFAP、Iba1(总或活化)和CD68免疫染色的SNpc切片被量化为每个标记物在同侧(注射)或对侧(未注射)半球占据的面积百分比。根据形态学标准,使用Halo (T)从总小胶质细胞中鉴定出活化的iba1阳性小胶质细胞。条形图代表平均值±SEM (n=5-8只/组)。经单因素方差分析,*P < 0.05或**P < 0.01
在此,我们研究了VPS35和αSyn在细胞和鼠脑中的生化、功能和病理相互作用。我们未能确定VPS35和αSyn之间的强大的生化相互作用,以及αSyn变异体过表达对反转录蛋白水平的有限影响,而VPS35的缺失未能诱导αSyn在人类细胞中的积累。在小鼠大脑中,我们发现由病毒介导的人D620N VPS35表达引起的黑质纹状体多巴胺能通路的强烈变性并没有被内源性αSyn的缺失所强烈改变。D620N VPS35的表达确实会导致黑质中pSer129-αSyn和全长APP免疫反应性的增加,但这种影响在脑提取物中尚未得到证实,这可能意味着这些标记物的重新分配而不是它们的积累。在一个发育致死性神经退行性表型的人类A53T-αSyn转基因小鼠模型中,我们没有发现αSyn病理影响的多个脑区存在物理逆转录酶缺乏的证据。此外,VPS35杂合性对A53T-αSyn模型中发生的αSyn神经病理或致死性神经退行性表型没有影响。最后,我们发现病毒介导的人WT VPS35过表达对大鼠黑质中人WT αSyn过表达诱导的黑质纹状体多巴胺能通路变性、αSyn病理或反应性胶质瘤没有保护作用。我们的数据表明,内源性αSyn表达并不是人D620N VPS35诱导的多巴胺能神经退行性变所必需的,此外,我们没有发现αSyn突变体诱导逆转录蛋白缺乏或恢复VPS35足以介导αSyn诱导的神经毒性的神经保护。综上所述,我们的研究未能为VPS35和αSyn在介导啮齿动物大脑神经变性中的功能或病理双向相互作用提供令人信服的支持,这表明这两种蛋白可能通过不同的途径起作用。
最近对种系杂合子VPS35缺失的小鼠模型的研究[22],或在黑质多巴胺能神经元中选择性地条件纯合子VPS35缺失的小鼠模型[25]表明,VPS35水平的降低会导致神经元中总αSyn的积累。然而,由pSer129-αSyn或αSyn聚集所定义的αSyn病理在这些敲除模型的大脑中尚未报道[22,25]。同样,D620N VPS35敲入小鼠不表现出内源性αSyn病理[30],也没有能力调节人类A53T-αSyn转基因小鼠的致死性神经退行性表型[30],但有证据表明,大脑中总αSyn的积累是适度的[31]。尽管没有明显的αSyn聚集,但有研究表明,在这些敲除或敲入模型中,神经元αSyn的积累可能导致神经变性[22,25,31]。我们之前报道过aav介导的人D620N VPS35在成年大鼠黑质中的表达足以诱导多巴胺能神经元的变性,但这是在没有明显αSyn病理的情况下发生的[32]。在这里,我们将这些研究扩展到小鼠,以解决内源性αSyn介导D620N VPS35致病作用的功能需求。虽然我们发现D620N VPS35可以特异性诱导WT小鼠同侧黑质中pSer129-αSyn的免疫反应性(图3A),而在SNCA KO小鼠中不存在,从而证实了该信号的特异性,但在该AAV模型中,我们没有观察到αSyn去除对多巴胺能神经元损失的强大影响(图2)。我们的研究表明,虽然VPS35可以调节神经元体和中脑腹侧过程中的pSer129-αSyn水平,这对D620N VPS35的退行性作用没有意义,因此它可能是良性的,不代表一个关键的下游致病事件。现在需要进一步的研究VPS35敲除或D620N VPS35敲除小鼠与SNCA KO小鼠杂交,以确定内源性αSyn是否有助于这些模型中出现的神经退行性表型。
目前尚不清楚VPS35缺失或pd相关突变如何在这些小鼠模型中驱动αSyn的积累。由于αSyn不是一个已知retromer货物和我们的研究在人类细胞中不支持两种蛋白质的物理相互作用(图1)。这是最有可能的一个VPS35对αSyn积累的影响是间接的,可能造成损害的溶酶体降解由于异常retromer排序CI-M6PR交付溶酶体酶等的溶酶体组织蛋白酶D腔[43]20日,21日,或通过CMA受体受损的排序,LAMP2A[22]。虽然调节VPS35可以适度调节大脑中αSyn的水平[22,25,31,43],但不表现出经典的αSyn病理,但VPS35已被令人信服地证明可以调节易聚集的微管相关蛋白tau[19]。一些研究表明,在人类tau病(进行性核上性麻痹和匹克病)和阿尔茨海默病的受影响脑区,逆转录亚基蛋白水平降低[44,45],沉默大脑中VPS35的表达可加剧人类P301S-tau转基因小鼠的tau神经病理以及运动和学习障碍[45]。相反,在3xTg AD小鼠模型中,使用药物伴侣TPT-172 (R33)的逆转录物稳定可以减少Aβ沉积和异常tau,并改善记忆障碍[46]。在PD-linked D620N VPS35敲入小鼠中,显示缺乏αSyn病理,相反,这些小鼠在大脑中表现出异常体突tau的广泛积累,其特征是病理性过磷酸化和构象特异性表位[30]。因此,尽管VPS35似乎具有调节多种蛋白质聚集途径的能力[19],但我们的观察表明,αSyn的积累可能在PD的啮齿动物模型中不起关键作用。在未来的研究中,类似地解决tau病理对pd相关VPS35突变在此类啮齿动物模型中诱导的神经变性的贡献将是重要的。
多项研究表明,在酵母、秀丽隐杆线虫和果蝇PD模型中,降低VPS35表达可加重人α - syn相关的毒性[27,28]。目前尚不清楚减少VPS35的作用是特定于α - syn依赖途径,还是更普遍地作用于降低细胞健康和活力。其他研究表明,增加小鼠海马神经元中VPS35的表达足以介导表达人WT αSyn的转基因小鼠的神经保护作用(line 61)[28]。这些研究结合起来倾向于支持人类αSyn诱导逆转录酶缺乏的概念,VPS35缺失加剧了αSyn的致病作用,而VPS35表达增加具有保护作用。这一机制与PD中易损神经元的关系尚不清楚。在我们尝试重建和扩展之前的研究中,我们发现,尽管αSyn病理负担很高,但与症状前或终末期疾病小鼠相比,人类A53T-αSyn转基因小鼠在受影响的中脑和后脑区域的逆转录亚基水平并未降低(图4)。与这一发现一致,种系杂合缺失VPS35并不会加剧αSyn的积累。这是A53T-αSyn小鼠模型的特征(图5)。与先前研究的一个关键区别是,在这些较低生物中纯合子VPS35缺失并不像在小鼠中那样致命,因此我们的研究仅限于使用杂合子VPS35缺失的小鼠,这些小鼠可能不足以加剧A53T-αSyn的致病作用。虽然我们的数据不支持在低等生物中报道的逆转录物可能作用于αSyn下游的发现,但在我们的小鼠研究中,可能没有充分捕捉到最相关的神经元群或大脑区域。人类A53T-αSyn转基因小鼠发生肢体瘫痪和生存受损主要是由于脊髓运动神经元的退化,但这些小鼠确实表现出广泛的神经元αSyn病理[34,47],然而,我们没有评估前脑结构或特定神经元群(如黑质多巴胺能神经元)中的逆转录物水平。然而,我们没有发现证据表明人类A53T-αSyn在这种特殊的转基因小鼠模型中驱动物理或功能上的逆转录缺陷。确定基于人类WT αSyn(即61号系)的转基因模型或由内源性(即BAC小鼠)而不是异位启动子驱动的转基因模型是否在相关大脑区域的反转录亚基水平上显示出改变,将是一项有趣的研究。
我们进一步扩展了先前的研究,报道了VPS35过表达对转基因小鼠海马WT αSyn的保护作用[28],建立了基于aav介导的人WT αSyn传递的具有良好特征和强大功能的PD大鼠模型[37,41]。这使我们能够直接评估VPS35在黑质纹状体多巴胺能通路中的神经保护作用。我们发现过表达WT VPS35对人WT αSyn表达诱导的神经多巴胺能神经元损失、αSyn病理和反应性胶质细胞形成的影响很小(图6、7和8)。在我们的αSyn大鼠模型中,WT VPS35缺乏保护作用可能反映了不同物种(大鼠与小鼠)之间的差异,不同神经元群体(神经多巴胺能神经元与海马锥体神经元)的易感性,或者转基因和病毒介导的人α - syn表达之间的内在技术或机制差异。例如,VPS35在啮齿动物海马神经元中被显著检测到,在那里它与tau病理的积累有关[30,39,44,45]。值得注意的是,我们确实发现D620N VPS35对α syn诱导的神经毒性具有适度的保护作用(图6D-G),这支持了这种pd相关突变可能通过功能获得机制(由于活性增加或改变)起作用的概念[19,48]。虽然我们在该模型中确实观察到D620N VPS35诱导的神经αSyn病理增加(图8),但这种效应很可能与该组多巴胺能神经元损失水平较低有关(图6E)。我们的研究表明,由人WT αSyn诱导的pd相关神经元群的变性很可能不是由于VPS35表达不足。有可能多巴胺能神经元在αSyn表达后确实降低了逆转录活性,但这需要恢复多个货物选择复合物的亚基。未来的研究可能会通过对这些啮齿动物模型给予R55或R33等药物伴侣来解决反转录物在α syn诱导的神经毒性中的作用[49],以稳定和增加整个反转录物复合物而不是单个亚基。我们的研究在证明仅恢复WT VPS35水平不足以保护多巴胺能神经元免受人类α syn诱导的神经毒性方面发挥了重要作用,这与海马锥体神经元所报道的作用不同[28]。
总的来说,我们的数据不能提供强有力的支持VPS35和αSyn之间的双向通路在多个具有良好特征的啮齿动物PD模型中。我们证明内源性αSyn对小鼠D620N VPS35的神经退行性作用并不是必需的,并且通过缺失或过表达来调节VPS35的水平对两种不同且强大的人类αSyn诱导的啮齿动物模型(A53T-αSyn转基因小鼠或注射AAV-WT-αSyn的大鼠)的影响很小。为了进一步明确,未来的研究需要在死后的人类大脑中进行,以评估Lewy病理是否是VPS35-linked PD的主要特征[7],此外,SNCA-linked PD大脑在受影响区域或神经元中是否表现出与AD、tau病和ALS大脑中观察到的相似的反转录亚基减少[19,33,44,45]。


发表评论
暂时没有评论,来抢沙发吧~